| Citation: | Xiaran Miao, Jinyou Lin, Fenggang Bian. Utilization of discarded crop straw to produce cellulose nanofibrils and their assemblies[J]. Journal of Bioresources and Bioproducts, 2020, 5(1): 26-36. doi: 10.1016/j.jobab.2020.03.003

|
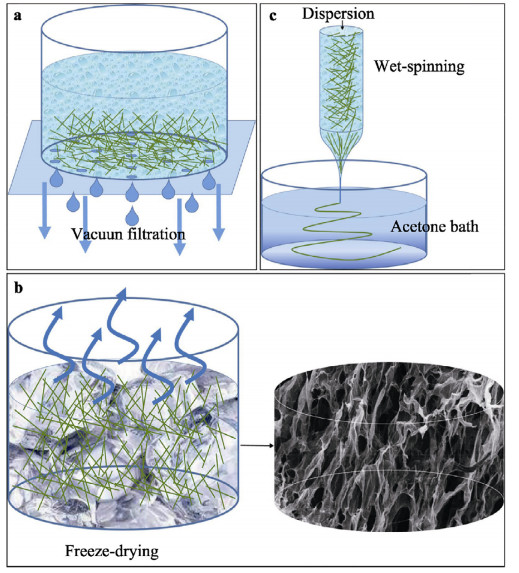
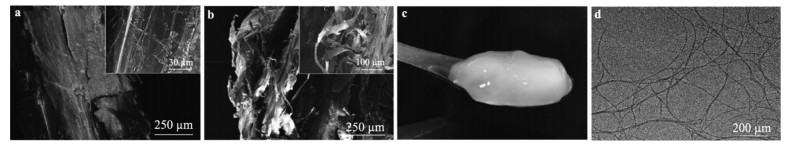
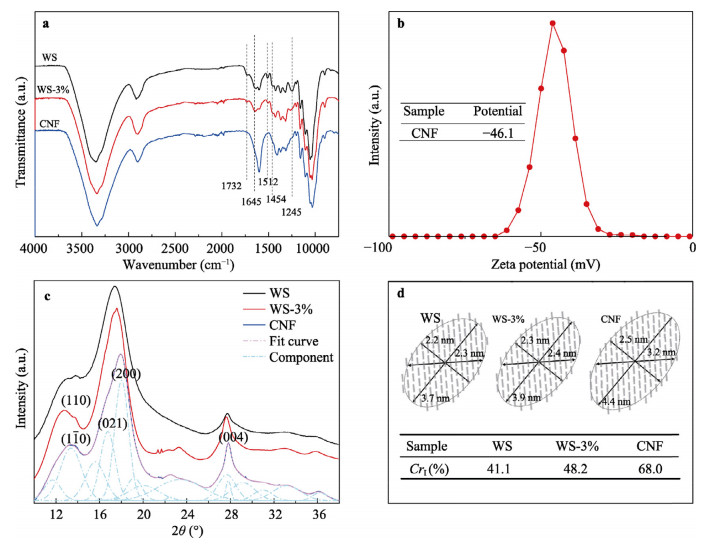
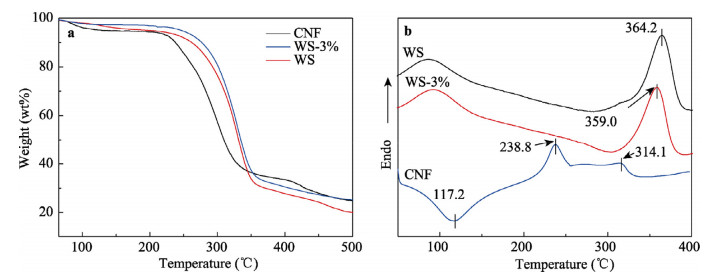
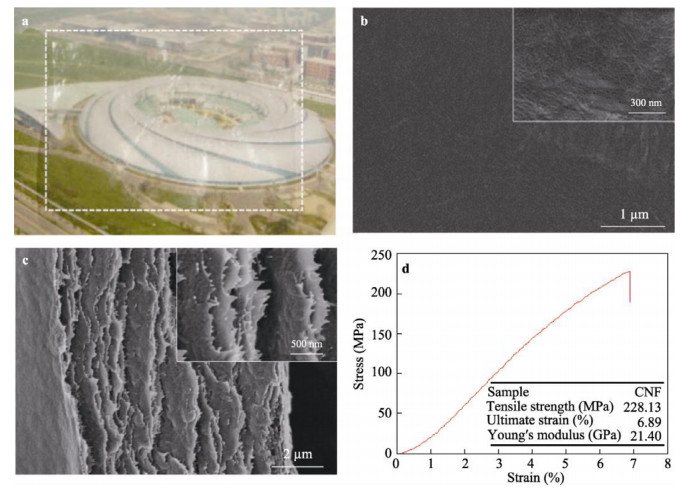
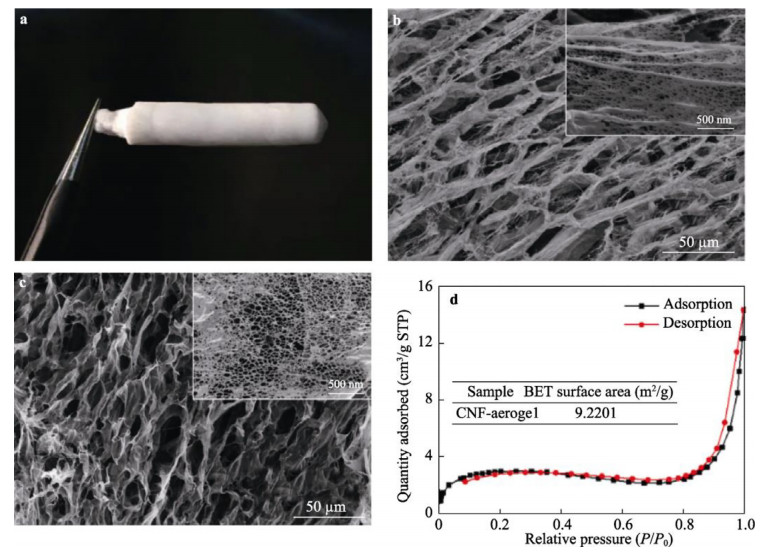
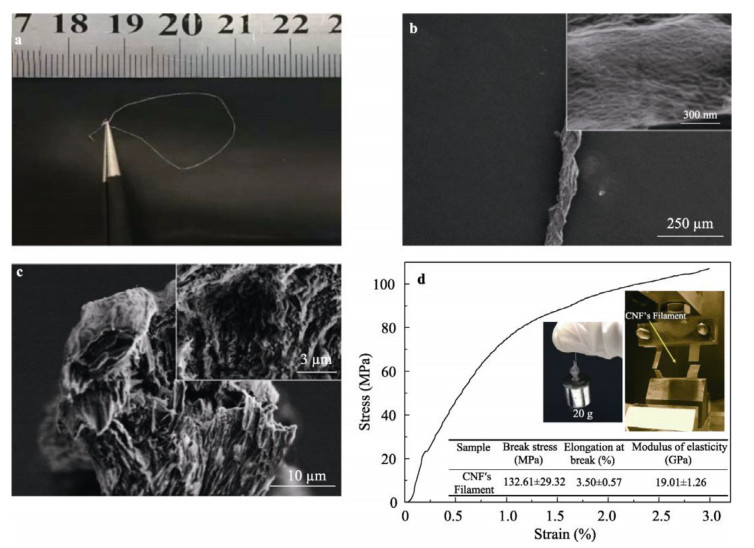
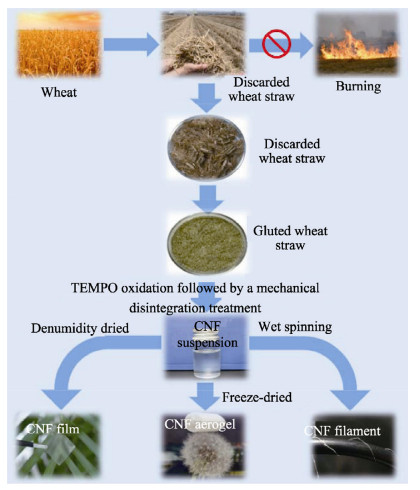
A tremendous amount of wheat straw (WS) has been generated by wheat crops every year, while only a small percentage is being used in applications, and most get burned on the field, causing a large amount of the exhaust gas that pollutes the environment. Herein, we report on the extraction of cellulose nanofibrils (CNF) from the alkali treated WS by a combination of TEMPO-oxidation and mechanical disintegration method. The crystalline structures, thermal properties, natural charge of the CNF were examined. The resultant nano-building blocks of CNF was assembled into macroscopic cellulose materials, i.e., film, aerogel, and filament in this work. Furthermore, the morphologies and microstructues as well as other properties of these three kinds of the CNF assemblies were investigated. The obtained CNF and its assemblies showed a potential application in new materials areas. This work explored a new way to utilize the discarded WS instead of being burned.
|
Alemdar, A., Sain, M., 2008. Biocomposites from wheat straw nanofibers:Morphology, thermal and mechanical properties. Compos. Sci. Technol. 68, 557-565. doi: 10.1016/j.compscitech.2007.05.044
|
|
Aulin, C., Salazar-Alvarez, G., Lindström, T., 2012. High strength, flexible and transparent nanofibrillated cellulose-nanoclay biohybrid films with tunable oxygen and water vapor permeability. Nanoscale 4, 6622.
|
|
Benítez, A.J., Torres-Rendon, J., Poutanen, M., Walther, A., 2013. Humidity and multiscale structure govern mechanical properties and defor-mation modes in films of native cellulose nanofibrils. Biomacromolecules 14, 4497-4506. doi: 10.1021/bm401451m
|
|
Bian, H.Y., Gao, Y., Yang, Y.Q., Fang, G.G., Dai, H.Q., 2018a. Improving cellulose nanofibrillation of waste wheat straw using the combined methods of prewashing, p-toluenesulfonic acid hydrolysis, disk grinding, and endoglucanase post-treatment. Bioresour. Technol. 256, 321-327. doi: 10.1016/j.biortech.2018.02.038
|
|
Bian, Z.X., Miao, X.R., Lin, J.Y., Tian, F., Bian, F.G., Li, H., 2018b. Extraction and structural investigation of jute cellulose nanofibers. Nucl. Sci. Tech. 29, 106. doi: 10.1007/s41365-018-0433-x
|
|
Carrillo, F., Colom, X., Suñol, J., Saurina, J., 2004. Structural FTIR analysis and thermal characterisation of lyocell and viscose-type fibres. Eur. Polym. J. 40, 2229-2234. doi: 10.1016/j.eurpolymj.2004.05.003
|
|
de France, K.J., Hoare, T., Cranston, E.D., 2017. Review of hydrogels and aerogels containing nanocellulose. Chem. Mater. 29, 4609-4631. doi: 10.1021/acs.chemmater.7b00531
|
|
Fukuzumi, H., Saito, T., Iwata, T., Kumamoto, Y., Isogai, A., 2009. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 10, 162-165. doi: 10.1021/bm801065u
|
|
Fukuzumi, H., Saito, T., Okita, Y., Isogai, A, 2010. Thermal stabilization of TEMPO-oxidized cellulose. Polym. Degrad. Stab. 95, 1502-1508. doi: 10.1016/j.polymdegradstab.2010.06.015
|
|
Han, J.Q., Zhou, C.J., Wu, Y.Q., Liu, F.Y., Wu, Q.L., 2013. Self-assembling behavior of cellulose nanoparticles during freeze-drying:effect of suspension concentration, particle size, crystal structure, and surface charge. Biomacromolecules 14, 1529-1540. doi: 10.1021/bm4001734
|
|
Isogai, A., Saito, T., Fukuzumi, H., 2011. TEMPO-oxidized cellulose nanofibers. Nanoscale. 3(1), 71-85. doi: 10.1039/C0NR00583E
|
|
Iwamoto, S., Isogai, A., Iwata, T., 2011. Structure and mechanical properties of wet-spun fibers made from natural cellulose nanofibers. Biom-acromolecules 12, 831-836. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=128b1532c4a6f37d5f0c25bfc8053345
|
|
Iwamoto, S., Kai, W.H., Isogai, A., Iwata, T., 2009. Elastic Modulus of single cellulose microfibrils from tunicate measured by atomic force microscopy. Biomacromolecules 10, 2571-2576. doi: 10.1021/bm900520n
|
|
Iwamoto, S., Nakagaito, A.N., Yano, H., 2007. Nano-fibrillation of pulp fibers for the processing of transparent nanocomposites. Appl. Phys. A 89, 461-466. doi: 10.1007/s00339-007-4175-6
|
|
Kasyapi, N., Chaudhary, V., Bhowmick, A.K., 2013. Bionanowhiskers from jute:Preparation and characterization. Carbohydr. Polym. 92, 1116-1123. doi: 10.1016/j.carbpol.2012.10.021
|
|
Kaushik, A., Singh, M., 2011. Isolation and characterization of cellulose nanofibrils from wheat straw using steam explosion coupled with high shear homogenization. Carbohydr. Res. 346, 76-85. doi: 10.1016/j.carres.2010.10.020
|
|
Klemm, D., Heublein, B., Fink, H.P., Bohn, A., 2005. Cellulose:fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 44, 3358-3393. doi: 10.1002/anie.200460587
|
|
Lin, J.Y., Yu, L.B., Tian, F., Zhao, N., Li, X.H., Bian, F.G., Wang, J., 2014. Cellulose nanofibrils aerogels generated from jute fibers. Carbohydr. Polym. 109, 35-43. doi: 10.1016/j.carbpol.2014.03.045
|
|
Liu, Q., Lu, Y., Aguedo, M., Jacquet, N., Ouyang, C., He, W.Q., Yan, C.R., Bai, W.B., Guo, R., Goffin, D., Song, J.Q., Richel, A., 2017. Isola-tion of high-purity cellulose nanofibers from wheat straw through the combined environmentally friendly methods of steam explosion, mi-crowave-assisted hydrolysis, and microfluidization. ACS Sustainable Chem. Eng. 5, 6183-6191. doi: 10.1021/acssuschemeng.7b01108
|
|
Liu, R.G., Yu, H., Huang, Y., 2005. Structure and morphology of cellulose in wheat straw. Cellulose 12, 25-34. doi: 10.1007/s10570-004-0955-8
|
|
Malho, J.M., Laaksonen, P., Walther, A., Ikkala, O., Linder, M.B., 2012. Facile method for stiff, tough, and strong nanocomposites by direct exfoliation of multilayered graphene into native nanocellulose matrix. Biomacromolecules 13, 1093-1099. doi: 10.1021/bm2018189
|
|
Miao, X.R., Lin, J.Y., Tian, F., Li, X.H., Bian, F.G., Wang, J., 2016a. Cellulose nanofibrils extracted from the byproduct of cotton plant. Carbo-hydr. Polym. 136, 841-850. doi: 10.1016/j.carbpol.2015.09.056
|
|
Miao, X.R., Tian, F., Lin, J.Y., Li, H., Li, X.H., Bian, F.G., Zhang, X.Z., 2016b. Tuning the mechanical properties of cellulose nanofibrils rein-forced polyvinyl alcohol composites via altering the cellulose polymorphs. RSC Adv. 6, 83356-83365. doi: 10.1039/C6RA14517E
|
|
Moon, R.J., Martini, A., Nairn, J., Simonsen, J., Youngblood, J., 2011. Cellulose nanomaterials review: structure, properties and nanocomposites. Chem. Soc. Rev. 40, 3941.
|
|
Morán, J.I., Alvarez, V.A., Cyras, V.P., Vázquez, A., 2008. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 15, 149-159. doi: 10.1007/s10570-007-9145-9
|
|
Mulyadi, A., Zhang, Z., Deng, Y.L., 2016. Fluorine-free oil absorbents made from cellulose nanofibril aerogels. ACS Appl. Mater. Interfaces 8, 2732-2740. doi: 10.1021/acsami.5b10985
|
|
Oun, A.A., Rhim, J.W, 2016. Isolation of cellulose nanocrystals from grain straws and their use for the preparation of carboxymethyl cellu-lose-based nanocomposite films. Carbohydr. Polym. 150, 187-200. doi: 10.1016/j.carbpol.2016.05.020
|
|
Pang, J.H., Wu, M., Zhang, Q.H., Tan, X., Xu, F., Zhang, X.M., Sun, R.C., 2015. Comparison of physical properties of regenerated cellulose films fabricated with different cellulose feedstocks in ionic liquid. Carbohydr. Polym. 121, 71-78. doi: 10.1016/j.carbpol.2014.11.067
|
|
Pelissari, F.M., do Amaral Sobral, P.J., Menegalli, F.C., 2014. Isolation and characterization of cellulose nanofibers from banana peels. Cellulose 21, 417-432. doi: 10.1007/s10570-013-0138-6
|
|
Peng, N., Wang, Y.F., Ye, Q.F., Liang, L., An, Y.X., Li, Q.W., Chang, C.Y., 2016. Biocompatible cellulose-based superabsorbent hydrogels with antimicrobial activity. Carbohydr. Polym. 137, 59-64. doi: 10.1016/j.carbpol.2015.10.057
|
|
Rosa, S.M.L., Rehman, N., de Miranda, M.I.G., Nachtigall, S.M.B., Bica, C.I.D., 2012. Chlorine-free extraction of cellulose from rice husk and whisker isolation. Carbohydr. Polym. 87, 1131-1138. doi: 10.1016/j.carbpol.2011.08.084
|
|
Saito, T., Nishiyama, Y., Putaux, J.L., Vignon, M., Isogai, A., 2006a. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 7, 1687-1691. doi: 10.1021/bm060154s
|
|
Saito, T., Nishiyama, Y., Putaux, J.L., Vignon, M., Isogai, A., 2006b. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 7, 1687-1691. doi: 10.1021/bm060154s
|
|
Sánchez, R., Espinosa, E., Domínguez-Robles, J., Loaiza, J.M., Rodríguez, A., 2016. Isolation and characterization of lignocellulose nanofibers from different wheat straw pulps. Int. J. Biol. Macromol. 92, 1025-1033. doi: 10.1016/j.ijbiomac.2016.08.019
|
|
Sehaqui, H., Ezekiel Mushi, N., Morimune, S., Salajkova, M., Nishino, T., Berglund, L.A., 2012. Cellulose nanofiber orientation in nanopaper and nanocomposites by cold drawing. ACS Appl. Mater. Interfaces 4, 1043-1049. doi: 10.1021/am2016766
|
|
Sinha, E., Rout, S.K., 2009. Influence of fibre-surface treatment on structural, thermal and mechanical properties of jute fibre and its composite. Bull. Mater. Sci. 32, 65-76. doi: 10.1007/s12034-009-0010-3
|
|
van den Berg, O., Capadona, J.R., Weder, C., 2007. Preparation of homogeneous dispersions of tunicate cellulose whiskers in organic solvents. Biomacromolecules 8, 1353-1357. doi: 10.1021/bm061104q
|
|
Wang, B.C., Benitez, A.J., Lossada, F., Merindol, R., Walther, A., 2016. Bioinspired mechanical gradients in cellulose nanofibril/polymer na-nopapers. Angew. Chem. Int. Ed. 55, 5966-5970. doi: 10.1002/anie.201511512
|
|
Xu, F., Shi, Y.C., Wang, D.H., 2012. Structural features and changes of lignocellulosic biomass during thermochemical pretreatments:a syn-chrotron X-ray scattering study on photoperiod-sensitive Sorghum. Carbohydr. Polym. 88, 1149-1156. doi: 10.1016/j.carbpol.2012.01.041
|
|
Xu, X.Z., Liu, F., Jiang, L., Zhu, J.Y., Haagenson, D., Wiesenborn, D.P., 2013. Cellulose nanocrystals vs. Cellulose nanofibrils:a comparative study on their microstructures and effects as polymer reinforcing agents. ACS Appl. Mater. Interfaces 5, 2999-3009. http://d.old.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ0229470238/
|
|
Yang, H.P., Yan, R., Chen, H.P., Lee, D.H., Zheng, C.G., 2007. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 86, 1781-1788. doi: 10.1016/j.fuel.2006.12.013
|
|
Yang, Huang, Q.B., Payne, G.F., Sun, R.C., Wang, X.H., 2019. A highly conductive, pliable and foldable Cu/cellulose paper electrode enabled by controlled deposition of copper nanoparticles. Nanoscale 11, 725-732. doi: 10.1039/C8NR07123C
|
|
Yu, L.B., Lin, J.Y., Tian, F., Li, X.H., Bian, F.G., Wang, J., 2014. Cellulose nanofibrils generated from jute fibers with tunable polymorphs and crystallinity. J. Mater. Chem. A 2, 6402.
|
|
Zhang, Y.X., Nypelö, T., Salas, C., Arboleda, J., Hoeger, I.C., Rojas, O.J., 2013. Cellulose nanofibrils. J. Renew. Mater. 1, 195-211. doi: 10.7569/JRM.2013.634115
|
Figures(8) / Tables(1)

Copyright © 2019 Editorial Office of Journal of Bioresources and Bioproducts
Supported by: Beijing Renhe Information Technology Co. Ltd support: info@rhhz.net



 DownLoad:
DownLoad: