| Citation: | HU Yun, JIA Puyou, SHANG Qianqian, ZHANG Meng, FENG Guodong, LIU Chengguo, ZHOU Yonghong. Synthesis and Application of UV-curable Phosphorous-Containing Acrylated Epoxidized Soybean Oil-based Resins[J]. Journal of Bioresources and Bioproducts, 2019, 4(3): 183-191. doi: 10.12162/jbb.v4i3.007

|
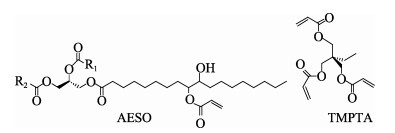
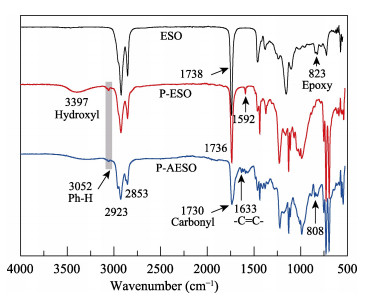
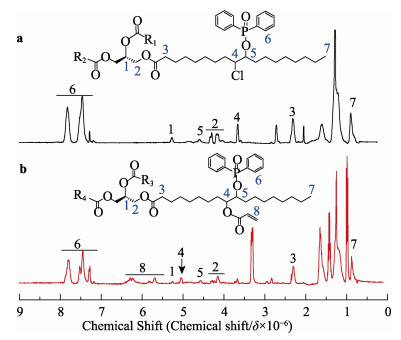
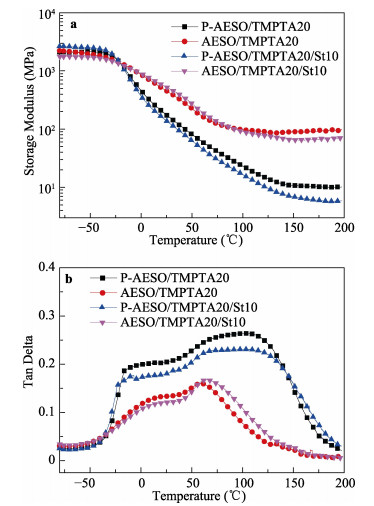
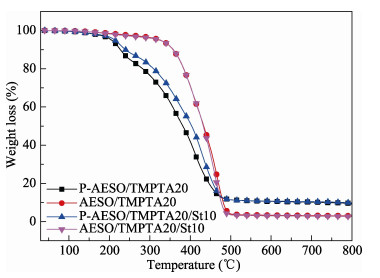
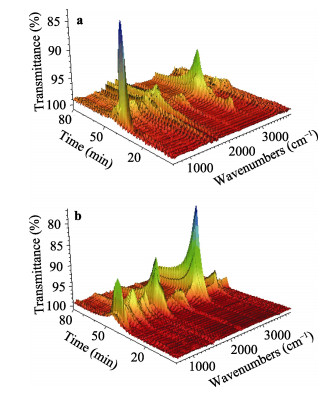
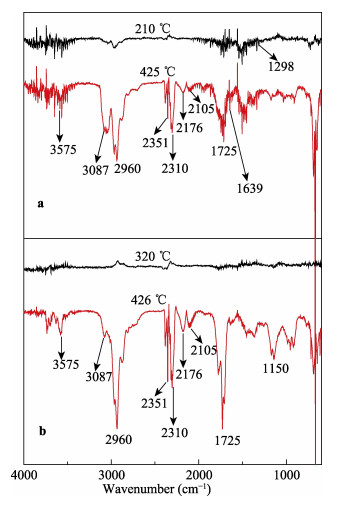
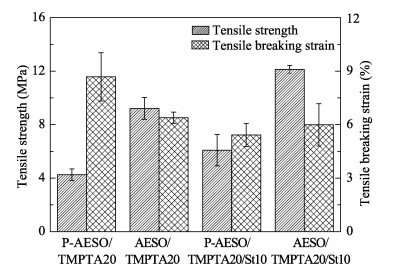
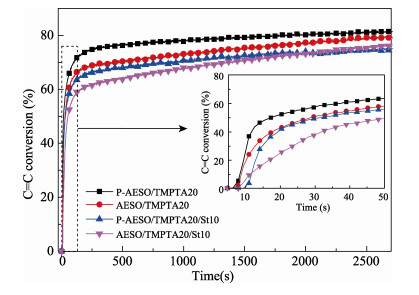
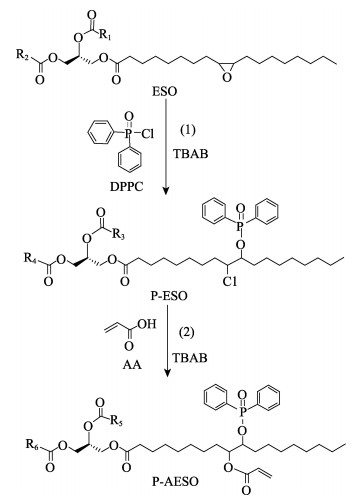
A novel phosphorous-containing acrylated epoxidized soybean oil-based (P-AESO) resin was developed via the ring-opening reaction of epoxidized soybean oil (ESO) with diphenylphosphinic chloride (DPPC), followed by acrylation of the resulting groups. The chemical structure was characterized by Fourier transform infrared spectroscopy (FT-IR), and 1H nuclear magnetic resonance (1H NMR). Subsequently, the viscosity and volumetric shrinkage of the obtained P-AESO resins were studied. Then the oligomer was formulated into UV-curable coatings, and the mechanical, thermal, and coating properties of the resulting UV-cured bioresins were studied by tensile testing, dynamic mechanical thermal analysis (DMA), thermogravimetric analysis (TGA) coupled with FT-IR spectroscopy (TGA-FT-IR), hardness, adhesion, pencil hardness and chemical resistance. Furthermore, the UV-curing behavior of the P-AESO resin was determined by real-time realtime infrared (RT-IR). Meanwhile, compared with coating from acrylated epoxidized soybean oil (AESO), the P-AESO system coatings showed better volumetric shrinkage, excellent adhesion, and enhanced thermal and glass transition temperature (Tg) while maintaining reasonably final C=C conversions and cross-link density. For instance, the obtained P-AESO/trimethylolpropanetriacrylate (TMPTA) 20 material possessed a volumetric shrinkage of 4.1%, Tg of 115.6℃, char yield of 9.47%, and final C=C conversions of 81.4% respectively, which exhibited superior values than that of the AESO/TMPTA20 material. The improvement of the P-AESO coating performances could contribute to the architectures that combined the structural features of phosphorous-containing rigid benzene. The developed P-AESO resin is promising for applications in the UV-curable coatings.
|
Auclair N, Kaboorani A, Riedl B, et al. 2015. Acrylated betulin as a comonomer for bio-based coatings. Part I:Characterization, photo-polymerization behavior and thermal stability. Industrial Crops and Products, 76:530-537. DOI: 10.1016/j.indcrop.2015.07.020.
|
|
Auclair N, Kaboorani A, Riedl B, et al. 2016. Acrylated betulin as a comonomer for bio-based coatings. Part Ⅱ:Mechanical and optical properties. Industrial Crops and Products, 82:118-126. DOI: 10.1016/j.indcrop.2015.11.081.
|
|
Chen X L, Huo L L, Jiao C M, et al. 2013. TG-FTIR characterization of volatile compounds from flame retardant polyurethane foams materials. Journal of Analytical and Applied Pyrolysis, 100: 186-191. DOI: 10.1016/j.jaap.2012.12.017.
|
|
Dai J Y, Jiang Y H, Liu X Q, et al. 2016a. Synthesis of eugenol-based multifunctional monomers via a thiol-ene reaction and preparation of UV curable resins together with soybean oil derivatives. RSC Advances, 6(22):17857-17866. DOI: 10.1039/c6ra01420h.
|
|
Dai J Y, Liu X Q, Ma S Q, et al. 2016b. Soybean oil-based UV-curable coatings strengthened by crosslink agent derived from itaconic acid together with 2-hydroxyethyl methacrylate phosphate. Progress in Organic Coatings, 97:210-215. DOI: 10.1016/j.porgcoat.2016.04.014.
|
|
Dai J Y, Ma S Q, Liu X Q et al. 2015a. Synthesis of bio-based unsaturated polyester resins and their application in waterborne UV-curable coatings. Progress in Organic Coatings, 78:49-54. DOI: 10.1016/j.porgcoat.2014.10.007.
|
|
Dai J Y, Ma S Q, Wu Y G, et al. 2015b. High bio-based content waterborne UV-curable coatings with excellent adhesion and flexibility. Progress in Organic Coatings, 87:197-203. DOI: 10.1016/j.porgcoat.2015.05.030.
|
|
Dai J Y, Ma S Q, Zhu L X, et al. 2017. UV-thermal dual cured anti-bacterial thiol-ene networks with superior performance from renewable resources. Polymer, 108:215-222. DOI: 10.1016/j.polymer.2016.11.068.
|
|
Dai J Y, Yang S M, Teng N, et al. 2018. Synthesis of eugenol-based silicon-containing benzoxazines and their applications as bio-based organic coatings. Coatings, 8(3):88. DOI: 10.3390/coatings8030088.
|
|
Feng G D, Hu L H, Ma Y, et al. 2018. An efficient bio-based plasticizer for poly (vinyl chloride) from waste cooking oil and citric acid:Synthesis and evaluation in PVC films. Journal of Cleaner Production, 189:334-343. DOI: 10.1016/j.jclepro.2018.04.085.
|
|
Hu Y, Liu C G, Shang Q Q, et al. 2018a. Synthesis and characterization of novel renewable Castor oil-based UV-curable polyfunctional polyurethane acrylate. Journal of Coatings Technology and Research, 15(1):77-85. DOI: 10.1007/s11998-017-9948-z.
|
|
Hu Y, Shang Q Q, Tang J J, et al. 2018b. Use of cardanol-based acrylate as reactive diluent in UV-curable Castor oil-based polyurethane acrylate resins. Industrial Crops and Products, 117:295-302. DOI: 10.1016/j.indcrop.2018.02.053.
|
|
Hu Y, Shang Q Q, Wang C N, et al. 2018c. Renewable epoxidized cardanol-based acrylate as a reactive diluent for UV-curable resins. Polymers for Advanced Technologies, 29(6):1852-1860. DOI: 10.1002/pat.4294.
|
|
Jia P Y, Feng G D, Bo C Y, et al. 2018. A composition of phosphaphenanthrene groups-containing Castor-oil-based phosphate plasticizer for PVC:Synthesis, characterization and property. Journal of Industrial and Engineering Chemistry, 60:192-205. DOI: 10.1016/j.jiec.2017.11.006.
|
|
Jia P Y, Hu L H, Zhang M, et al. 2016. TG-FTIR and TG-MS analysis applied to study the flame retardancy of PVC-Castor oil-based chlorinated phosphate ester blends. Journal of Thermal Analysis and Calorimetry, 124(3):1331-1339. DOI: 10.1007/s10973-015-5199-3.
|
|
Li K B, Shen Y D, Fei G Q, et al. 2015. Preparation and properties of Castor oil/pentaerythritol triacrylate-based UV curable waterborne polyurethane acrylate. Progress in Organic Coatings, 78:146-154. DOI:10.1016/j.porgcoat. 2014.09.012.
|
|
Li P, Ma S Q, Dai J Y, et al. 2017. Itaconic acid as a green alternative to acrylic acid for producing a soybean oil-based thermoset:synthesis and properties. ACS Sustainable Chemistry & Engineering, 5(1):1228-1236. DOI: 10.1021/acssuschemeng.6b02654.
|
|
Liu C G, Dai Y, Hu Y, et al. 2016a. Highly functional unsaturated ester macromonomer derived from soybean oil:synthesis and copolymerization with styrene. ACS Sustainable Chemistry & Engineering, 4(8):4208-4216. DOI: 10.1021/acssuschemeng.6b00700.
|
|
Liu C G, Shang Q Q, Jia P Y, et al. 2016b. Tung oil-based unsaturated Co-ester macromonomer for thermosetting polymers:synergetic synthesis and copolymerization with styrene. ACS Sustainable Chemistry & Engineering, 4(6):3437-3449. DOI: 10.1021/acssuschemeng.6b00466.
|
|
Liu C G, Wang C N, Hu Y, et al. 2018. Castor oil-based polyfunctional acrylate monomers:Synthesis and utilization in UV-curable materials. Progress in Organic Coatings, 121:236-246. DOI: 10.1016/j.porgcoat.2018.04.020.
|
|
Liu H J, Lu W C, Liu S J. 2018. Development of acrylated soybean oil-based UV-curable coatings with high impact strength from low viscosity oligomer. Journal of Applied Polymer Science, 135(3):45698. DOI: 10.1002/app.45698.
|
|
Liu R, Luo J, Ariyasivam S, et al. 2017. High biocontent natural plant oil based UV-curable branched oligomers. Progress in Organic Coatings, 105:143-148. DOI: 10.1016/j.porgcoat.2016.11.009.
|
|
Liu R, Zhang X P, Zhu J J, et al. 2015. UV-curable coatings from multiarmed cardanol-based acrylate oligomers. ACS Sustainable Chemistry & Engineering, 3(7):1313-1320. DOI: 10.1021/acssuschemeng.5b00029.
|
|
Ma S Q, Jiang Y H, Liu X Q, et al. 2014. Bio-based tetrafunctional crosslink agent from Gallic acid and its enhanced soybean oil-based UV-cured coatings with high performance. RSC Advances, 4(44):23036. DOI: 10.1039/c4ra01311e.
|
|
Qian X D, Song L, Hu Y, et al. 2011. Combustion and thermal degradation mechanism of a novel intumescent flame retardant for epoxy acrylate containing phosphorus and nitrogen. Industrial & Engineering Chemistry Research, 50(4):1881-1892. DOI: 10.1021/ie102196k.
|
|
Sharmin E, Zafar F, Akram D, et al. 2015. Recent advances in vegetable oils based environment friendly coatings:A review. Industrial Crops and Products, 76:215-229. DOI: 10.1016/j.indcrop.2015.06.022.
|
|
Wang X L, Chen L, Wu J N, et al. 2017. Flame-retardant pressure-sensitive adhesives derived from epoxidized soybean oil and phosphorus-containing dicarboxylic acids. ACS Sustainable Chemistry & Engineering, 5(4):3353-3361. DOI: 10.1021/acssuschemeng.6b03201.
|
|
Wu Q, Hu Y, Tang J J, et al. 2018. High-performance soybean-oil-based epoxy acrylate resins:"Green" synthesis and application in UV-curable coatings. ACS Sustainable Chemistry & Engineering, 6(7):8340-8349. DOI: 10.1021/acssuschemeng.8b00388.
|
|
Zhang C Q, Garrison T F, Madbouly S A, et al. 2017. Recent advances in vegetable oil-based polymers and their composites. Progress in Polymer Science, 71:91-143. DOI: 10.1016/j.progpolymsci.2016.12.009.
|
|
Zhang Y H, Li Y Z, Wang L W, et al. 2017. Synthesis and characterization of methacrylated eugenol as a sustainable reactive diluent for a maleinated acrylated epoxidized soybean oil resin. ACS Sustainable Chemistry & Engineering, 5(10):8876-8883. DOI: 10.1021/acssuschemeng.7b01673.
|
|
Zhou X, Li Y, Fang C Q, et al. 2015. Recent advances in synthesis of waterborne polyurethane and their application in water-based ink:A review. Journal of Materials Science & Technology, 31(7):708-722. DOI: 10.1016/j.jmst.2015.03.002.
|
Figures(10) / Tables(3)

Copyright © 2019 Editorial Office of Journal of Bioresources and Bioproducts
Supported by: Beijing Renhe Information Technology Co. Ltd support: info@rhhz.net



 DownLoad:
DownLoad: