| Citation: | Zhongfei YUAN, Hongjia LIN, Xueren QIAN, Jing SHEN. Converting a Dilute Slurry of Hollow Tube-like Papermaking Fibers into Phase-reversible, Self-healable, and Stretchable Hydrogels[J]. Journal of Bioresources and Bioproducts, 2019, 4(4): 214-221. doi: 10.12162/jbb.v4i4.011

|
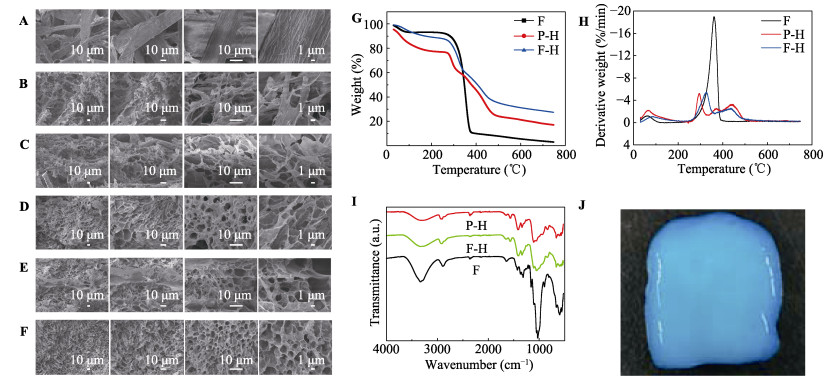
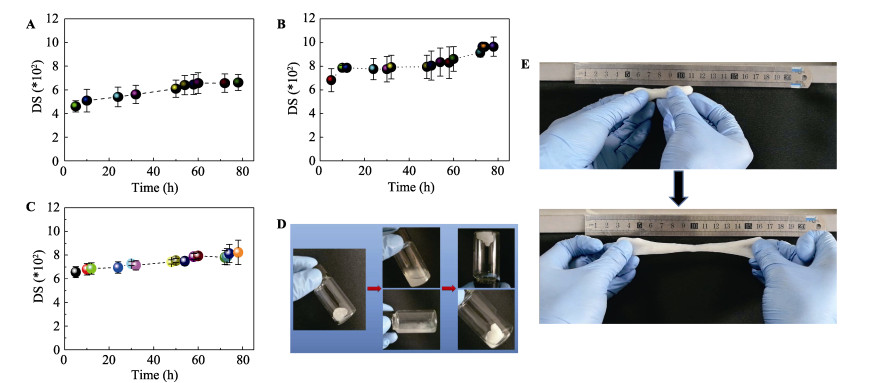
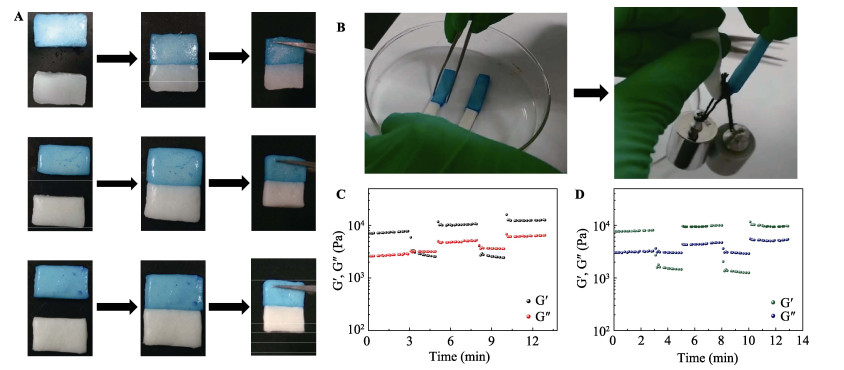
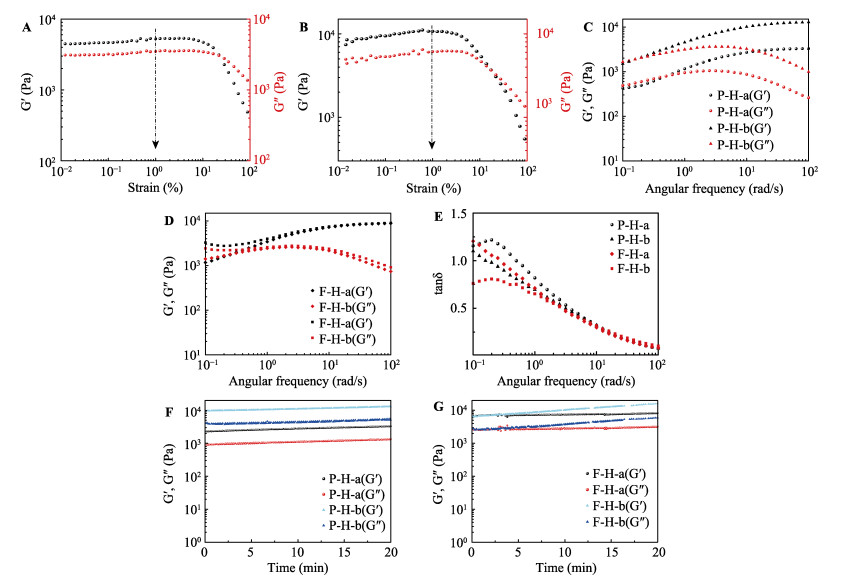
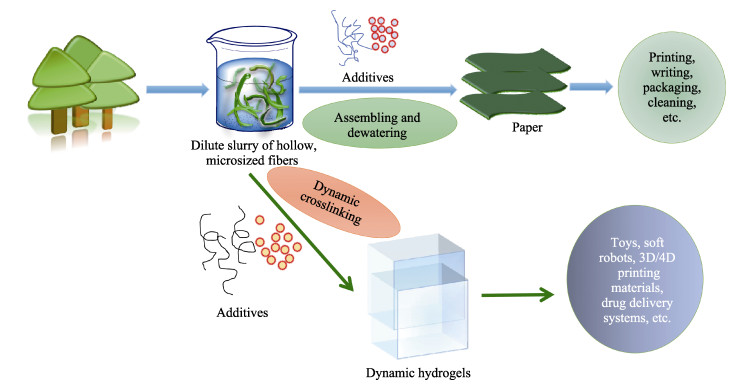
Commercially, assembly-directed packing of hollow tube-like papermaking fibers with diameters of roughly 10-50 μm into sustainable micro-fibrous bio-assemblies (i.e., cellulosic paper) starts with a dilute fiber slurry. In this process, a huge amount of water is required to disperse and transport fibers, which also facilitates colloidal interactions and formation of inter-fiber bonds. To form bio-assemblies in their dry states, unit operations associated with dewatering and drying are routine practices and treatment of the generated wastewater is a necessity. We herein present a facile, scalable concept of converting fiber slurry into dynamic hydrogels by using chemical additives (similar to papermaking wet-end additives), but without water removal. We used a typical group of additives as an example in an attempt to demonstrate the applicability of the concept. With boron-based dynamic chemistry as a key theoretical foundation, the combination of crosslinking and hydrogen bonding led to the formation of phase-reversible, self-healable, and stretchable hydrogels. Essentially, the characteristics of hydrogels can be facilely tunable, and process parameters such as polymer dosage are rather critical. It is worth noting that fibers can act as a structural skeleton or mechanical support for tailorable design of hydrogels. The concept demonstrated in this study would provide useful insights into value-added utilization of mass-producible biopolymeric fibers in accordance with existing mill facilities. Fiber-based hydrogels would find use in diversified applications:toys, 3D/4D printing materials, soft robots, drug delivery systems, among others.
|
Azevedo S, Costa A M S, Andersen A, et al., 2017. Bioinspired ultratough hydrogel with fast recovery, self-healing, injectability and cytocompatibility. Advanced Materials, 29(28):1700759.DOI: 10.1002/adma.201700759.
|
|
Bian H, Wei L, Lin C, et al., 2018. Lignin-containing cellulose nanofibril-reinforced polyvinyl alcohol hydrogels. ACS Sustainable Chemistry & Engineering, 6(4):4821-4828.DOI: 10.1021/acssuschemeng.7b04172.
|
|
Chakraborty P, Guterman T, Adadi N, et al., 2019. A self-healing, all-organic, conducting, composite peptide hydrogel as pressure sensor and electrogenic cell soft substrate. ACS Nano, 13(1):163-175. DOI: 10.1021/acsnano.8b05067.
|
|
Dikin D A, Stankovich S, Zimney E J, et al., 2007. Preparation and characterization of graphene oxide paper. Nature, 448(7152):457-460. DOI: 10.1038/nature06016.
|
|
Ding Q Q, Xu X W, Yue Y Y, et al., 2018. Nanocellulose-mediated electroconductive self-healing hydrogels with high strength, plasticity, viscoelasticity, stretchability, and biocompatibility toward multifunctional applications. ACS Applied Materials & Interfaces, 10(33):27987-28002. DOI: 10.1021/acsami.8b09656.
|
|
Fatehi P, Xiao H N, van de Ven T G M, 2011. Quantitative analysis of cationic poly(vinyl alcohol) diffusion into the hairy structure of cellulose fiber pores:charge density effect.Langmuir, 27(22):13489-13496. DOI: 10.1021/la203364x.
|
|
Fukahori S, Ichiura H, Kitaoka T, et al., 2003. Photocatalytic decomposition of bisphenol A in water using composite TiO2-zeolite sheets prepared by a papermaking technique.Environmental Science & Technology, 37(5):1048-1051.DOI: 10.1021/es0260115.
|
|
Han J Q, Lei T Z, Wu Q L, 2014. High-water-content mouldable polyvinyl alcohol-borax hydrogels reinforced by well-dispersed cellulose nanoparticles:dynamic rheological properties and hydrogel formation mechanism. Carbohydrate Polymers, 102:306-316. DOI: 10.1016/j.carbpol.2013.11.045.
|
|
Hong S H, Kim S, Park J P, et al., 2018. Dynamic bonds between boronic acid and alginate:hydrogels with stretchable, self-healing, stimuli-responsive, remoldable, and adhesive properties. Biomacromolecules, 19(6):2053-2061. DOI: 10.1021/acs.biomac.8b00144.
|
|
Ke H, Yang L P, Xie M, et al., 2019. Shear-induced assembly of a transient yet highly stretchable hydrogel based on pseudopolyrotaxanes. Nature Chemistry, 11(5):470-477.DOI: 10.1038/s41557-019-0235-8.
|
|
Li Z Q, Wang G N, Wang Y G, et al., 2018. Reversible phase transition of robust luminescent hybrid hydrogels. Angewandte Chemie, 130(8):2216-2220. DOI: 10.1002/ange.201712670.
|
|
Liu A D, Walther A, Ikkala O, et al., 2011. Clay nanopaper with tough cellulose nanofiber matrix for fire retardancy and gas barrier functions. Biomacromolecules, 12(3):633-641. DOI: 10.1021/bm101296z.
|
|
Lu B L, Lin F C, Jiang X, et al., 2017. One-pot assembly of microfibrillated cellulose reinforced PVA-borax hydrogels with self-healing and pH-responsive properties. ACS Sustainable Chemistry & Engineering, 5(1):948-956. DOI: 10.1021/acssuschemeng.6b02279.
|
|
Mansur H S, Sadahira C M, Souza A N, et al., 2008. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Materials Science and Engineering:C, 28(4):539-548. DOI: 10.1016/j.msec.2007.10.088.
|
|
Nojoomi A, Arslan H, Lee K, et al., 2018. Bioinspired 3D structures with programmable morphologies and motions.Nature Communications, 9:3705. DOI: 10.1038/s41467-018-05569-8.
|
|
Piest M, Zhang X L, Trinidad J, et al., 2011. pH-responsive, dynamically restructuring hydrogels formed by reversible crosslinking of PVA with phenylboronic acid functionalised PPO-PEO-PPO spacers (Jeffamines®). Soft Matter, 7(23):11111. DOI: 10.1039/c1sm06230a.
|
|
Rauner N, Meuris M, Zoric M, et al., 2017. Enzymatic mineralization generates ultrastiff and tough hydrogels with tunable mechanics. Nature, 543(7645):407-410. DOI: 10.1038/nature21392.
|
|
Sadri B, Goswami D, Sala de Medeiros M, et al., 2018.Wearable and implantable epidermal paper-based electronics.ACS Applied Materials & Interfaces, 10(37):31061-31068.DOI: 10.1021/acsami.8b11020.
|
|
Spoljaric S, Salminen A, Luong N D, et al., 2014. Stable, self-healing hydrogels from nanofibrillated cellulose, poly(vinyl alcohol) and borax via reversible crosslinking. European Polymer Journal, 56:105-117. DOI:10.1016/j.eurpolymj. 2014.03.009.
|
|
Sun J Y, Zhao X, Illeperuma W R K, et al., 2012. Highly stretchable and tough hydrogels. Nature, 489(7414):133.DOI: 10.1038/nature11409.
|
|
Tayeb A H, Hubbe M A, Tayeb P, et al., 2017. Soy proteins as a sustainable solution to strengthen recycled paper and reduce deposition of hydrophobic contaminants in papermaking:a bench and pilot-plant study. ACS Sustainable Chemistry & Engineering, 5(8):7211-7219. DOI:10.1021/acssuschemeng. 7b01425.
|
|
Taylor D L, in het Panhuis M, 2016. Self-healing hydrogels.Advanced Materials, 28(41):9060-9093. DOI: 10.1002/adma.201601613.
|
|
Tejado A, van de Ven T G M, 2010. Why does paper get stronger as it dries? Materials Today, 13(9):42-49. DOI: 10.1016/s1369-7021(10)70164-4.
|
|
Wang Q G, Mynar J L, Yoshida M, et al., 2010. High-watercontent mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature, 463(7279):339-343. DOI: 10.1038/nature08693.
|
|
Wu D, Li L M, Wang Y, et al., 2018. Localized liquefaction coupled with rapid solidification for miniaturizing/nanotexturizing microfibrous bioassemblies into robust, liquid-resistant sheet. ACS Sustainable Chemistry & Engineering, 6(11):15697-15707. DOI: 10.1021/acssuschemeng.8b04215.
|
|
Yang H P, Yan R, Chen H P, et al., 2007. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, 86(12/13):1781-1788. DOI: 10.1016/j.fuel.2006.12.013.
|
Figures(5)

Copyright © 2019 Editorial Office of Journal of Bioresources and Bioproducts
Supported by: Beijing Renhe Information Technology Co. Ltd support: info@rhhz.net



 DownLoad:
DownLoad: